1. 进化树介绍
参考:
https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogenetics
- 1. 进化树介绍
1.1. 系统发生学是什么
系统发生学是研究生物实体(通常是物种、个体或基因)之间进化关系的学科。系统发育的主要要素总结如下图1所示。
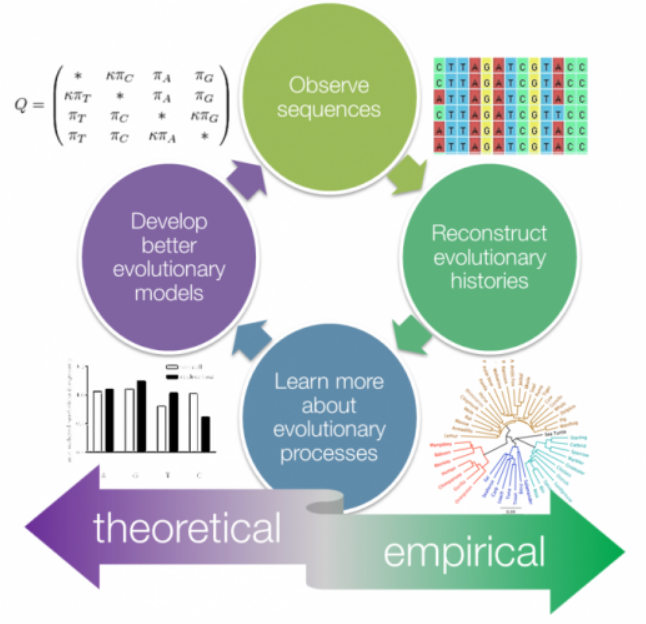
通常,系统发育学家研究以下类型的问题之一:
•我感兴趣的物种/个体/基因之间的进化关系或历史是什么?
•序列如何演变?
•我可以用数学模型更好地描述序列进化的过程吗?
我们可以通过查看核苷酸或蛋白质序列,并将其与我们对序列进化的理解(用进化模型来描述)相结合,来重建系统发育树。
这使我们能够推断过去发生的进化事件,并且还提供有关在序列上运行的进化过程的更多信息。
因此,我们可以加深对进化工作原理的理解,并开发出更好的进化数学模型。
1.2. Why use molecular data
今天,几乎所有的进化关系都可以从分子序列数据中推断出来。 这是因为:
•DNA是遗传物质;
•我们现在可以轻松,快速,廉价且可靠地对遗传材料进行测序;
•序列是高度特定的,通常信息丰富。
在极少数情况下,如果无法获得遗传物质(例如,某些古代化石样本),则可以使用形态学测量来推断进化关系。 但是,这种方法不如使用分子数据可靠,因为我们知道有时相同的形态性状可能来自多个独立的进化谱系(即该性状的发生是相似的)
( arise from multiple independent evolutionary lineages ) i.e. occurances of the trait are analogous
Analogous traits
Analogous traits 执行相似的功能,但具有不同的进化起源,即它们不具有共同的祖先
1.3. Applications of phylogenetics
系统发育学很重要,因为它丰富了我们对基因,基因组,物种(以及更普遍的分子序列)如何进化的理解。 通过系统发育学,我们不仅了解序列如何成为今天的样子,而且了解使我们能够预测它们将来如何变化的一般原理。 这不仅具有根本的重要性,而且对于许多应用都非常有用(图2)。
下面是一些可能的应用:
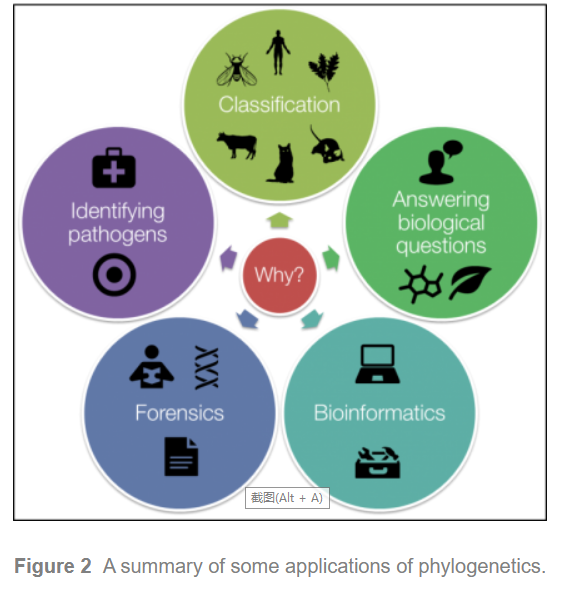
classification:基于序列数据的系统发育为我们提供了比分子测序之前更准确的亲缘模式描述。系统发育学现在为林奈新种分类提供了信息。
法医学(forensics):系统发育学是用来评估在法庭案件中提供的DNA证据,以告知情况,例如某人在哪里犯罪,什么时候食物被污染了,或者在哪里孩子的父亲是未知的。
确定病原体的来源:分子测序技术和系统发育方法可用于了解新的病原体暴发的更多信息。这包括查明病原体与哪些物种相似,以及随后可能的传播源。这可能导致对公共卫生政策的新建议。
Conservation :当保护生物学家必须做出艰难的决定以避免哪些物种灭绝时,系统遗传学可以帮助制定保护政策。
生物信息学和计算:许多为系统发生学开发的算法已被用于其他领域的软件开发。
其他
随着更新、更快的测序技术的出现,现在有可能把一台测序机带到野外,在原地对感兴趣的物种进行测序。需要系统发育来增加数据的生物学意义。
1.4. 系统发生树是什么
系统发生树,是一种解释序列是如何进化的,它们的系谱关系,因此它们是如何成为今天的样子。
A phylogeny, also known as a tree, is an explanation of how sequences evolved, their genealogical relationships, and therefore how they came to be the way they are today.
Genealogical
The direct descent from an ancestor, lineage or pedigree
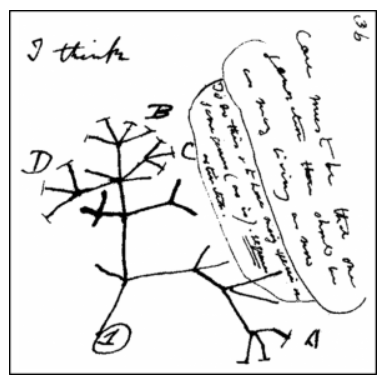
系统发育树的第一幅草图之一是查尔斯·达尔文(Charles Darwin)绘制的(图3)。
这张著名的图是他用来发展这个想法的一系列笔记中的早期素描之一。
1.5. 家系树
这是一个大家都熟悉的例子——一个家谱(图4)。你可能已经知道如何解释家谱上的亲缘关系模式,结果发现,同样的原则更普遍地适用于系统发育树。
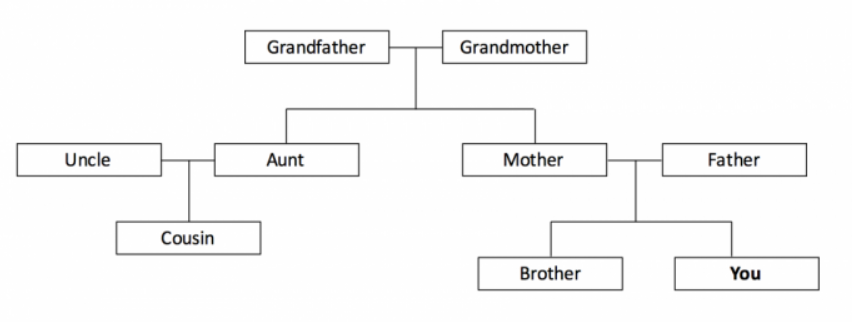
我们可以看到您与兄弟的关系比与您堂兄的关系更紧密。 将树追溯到您的祖先,您会看到原因。 您与兄弟共享的最新共同祖先(MRCA)是您的母亲。 相比之下,您与第一个堂兄共享的MRCA是您的外祖母。
当您阅读更复杂的系统发育史时,这些原则也适用-请记住追溯到MRCA,以检查相关性模式。
1.6. 树的解释
https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogeny/aspects-phylogenies/topology
https://artic.network/how-to-read-a-tree.html
https://science.sciencemag.org/content/310/5750/979
具体来说,我们将专注于:
(1)拓扑
(2)分支
(3)节点
(i)提示
(ii)内部节点
(iii)根
(4)置信度
1.6.1. 拓扑结构
拓扑结构是树的分支结构。
它具有特殊的生物学意义,因为它表明了taxa的亲缘关系模式,这意味着具有相同拓扑结构和root的树
具有相同的生物学解释。
meaning that trees with the same topology and root have the same biological interpretation.
例子:
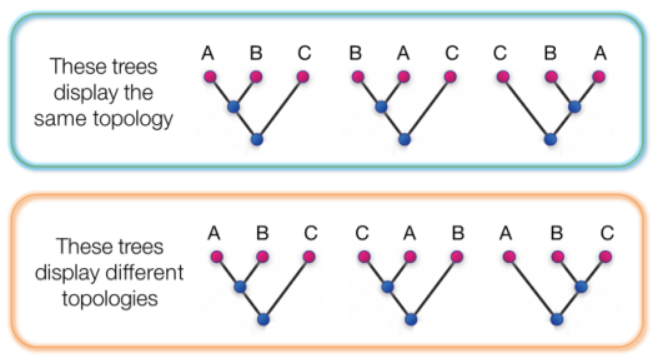
顶部框中的三棵树具有相同的拓扑。
这一点从“A和B之间的关系比它们和C之间的关系更密切”这句话中可以看出,这对所有三棵树都适用。
在下面的框中,所有三棵树都具有不同的拓扑结构。
例如,在最左侧的树,A和B更密切相关的另一个比C。
然而,在中间的树,它是C和彼此的近亲,在右边树是B和C是最密切相关的。
1.6.2. 分支
分支显示了遗传信息从一代传给下一代的途径。
分支长度代表了遗传变化,即分支越长,积累的遗传变化( divergence )越多。
Genetic change = Evolutionary rate x Divergence time
(substitutions/site) (substitutions/site/year) (years)
通常,我们通过估计每个位点的核苷酸或蛋白质取代的平均数来衡量遗传变化的程度。
例子:
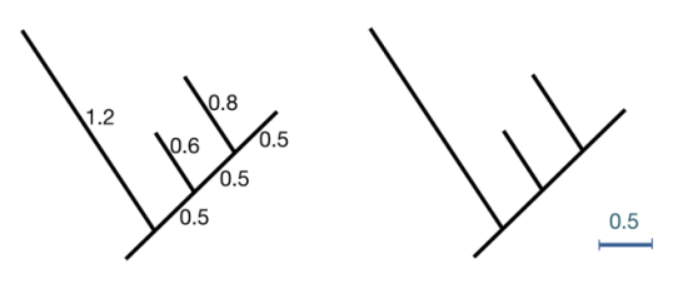
分支的长度通常按比例绘制,并指出每个位点的取代数。
系统发育偶尔会显示分支长度(左),但是用比例尺表示的分支长度(右)更为常见。
因此,将尺子放在手边以解释您在文献中看到的系统发育很有用!
- 如何估计遗传变化?
估计遗传变化的程度不是一件小事。
一种简单的方法是align序列对,计算差异的个数,然后除以序列长度。

在上面的简单比对中,我们可以看到两个序列之间有一个位点不同,我们可以说,根据这个小样本,每个位点有1/10 = 0.1个替换。
然而,这是假设每一个已经发生的都被我们观察到,因此不考虑任何多重替换。
还需要假设每一次替换(例如从T>C,或>G)都是等可能发生的,现在我们知道这是不现实的。
为了克服这些问题,现在普遍使用进化模型来推断已经发生的遗传变化。
- Beware of very long branches!
要使用上述简单方法获得每个位点都存在一个替换的情况,将要求一对序列在所有位点(10/10)上彼此完全不同。
您不太可能比对此类序列,因为两个随机核苷酸序列很可能具有25%的同一性。
因此,如果您在文献中看到每个站点的分支长度超过〜3个替换的数字,那么您可能要担心我们对这些估计的信心!
1.6.3. 节点
更多基因树与物种树的差别,查看: https://www.cnblogs.com/bingoup/p/11112360.html
节点是分支末端的点,代表进化历史中各个点的序列或假设序列。
下图中显示了三种类型的节点及其在示例系统发育中的位置。
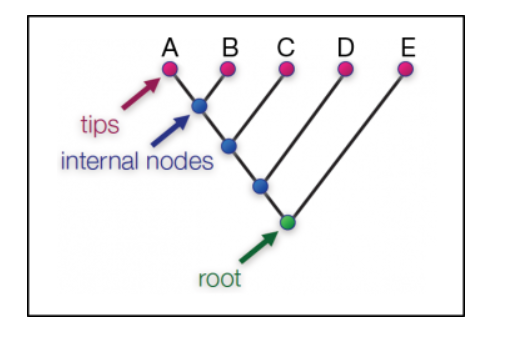
1.tips
tips上每一个点代表了我们进行系统发育树推断所用的一条真实序列,
用于构建系统发育的序列出现在单个末端分支上,称为末端或外部节点。
2.内部节点
内部节点出现在多个分支相遇的点处,并代表(通常是推断的)祖先序列。
例如,在图中,蓝色箭头指示的内部节点是序列A和B的假设共同祖先。
如果进化树是一颗基因树(多基因族构建树),那么internal nodes代表了基因复制事件;如果进化树是一颗物种树,那么内部节点代表了一个物种形成事件。
多基因族 multigene family
多基因族 (multigene family )是真核生物基因结构的特征之一,是由某一祖先基因经过重复和变异所产生的一组基因。大致可分为两类:一类是基因家族成簇地分布在某一条染色体上,它们可同时发挥作用,合成某些蛋白质;另一类是一个基因家族的不同成员成簇地分布不同染色体上,这些不同成员编码一组功能上紧密相关的蛋白质。多基因族是基因重复的最显著的特征。
基因复制事件:
基因复制(或染色体复制或基因扩增)是在分子进化过程中产生新遗传物质的主要机制。
可以将其定义为包含基因的DNA区域的任何重复。
基因复制可能是DNA复制和修复机制中几种错误类型的产物,也可能是由于自私的遗传元件的偶然捕获而引起的。
基因复制的常见来源包括异位重组,逆转座事件,非整倍性,多倍性和复制滑移。
3.根
根是一个非常重要的内部节点,代表着系统发育中所有序列的最近共同祖先。 我们将在下一节详细讨论根…
1.6.4. 根
根是树中所有分类单元最近的共同祖先。
因此,它是这棵树最古老的部分,它告诉我们进化的方向,随着遗传信息的流动,从根部流向tips,每一代都是如此。
大多数系统发育重建的方法都不能估计根的位置,部分原因是这增加了可能的树的数量,从而增加了计算树的时间。
下图显示了一个unrooted tree(左)的例子,它随后被rooted(右)。
注意左图中,root的位置是未知的。但有一些算法可以推测出来。
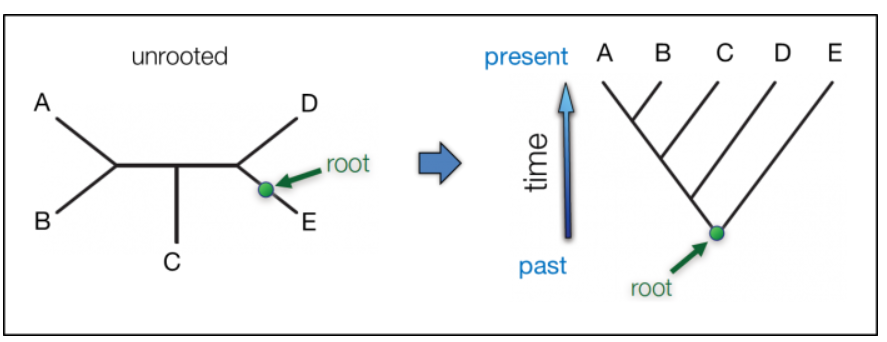
- Rooting a tree affects its meaning
确定正确的根位置对于系统发育解释至关重要,因为根可以告诉我们进化的方向,因此会影响我们对亲缘性模式所做的陈述。
because the root tells us the direction of evolution and so affects statements that we make about patterns of relatedness.
例如,在上面的无根树(左)中,如果root的位置未知,我们无法做出诸如“ A与B的关系比与C的关系更紧密”之类的语句,因为如果根出现在连接 A和B的分支之间的任何位置,那将是不正确的。
- 估计根位置的方法
1.Outgroup rooting
使用外群是首选的方法。
在分析中纳入一个或多个“外群”序列,这些序列与我们感兴趣的序列之间的距离比序列彼此之间的距离更远。 这些序列通常称为“外群”。
因此,根估计的位置就是“外群”加入树的其余部分的点。
**“外群”的最好选择是与我们感兴趣的序列最密切相关的可用分组。 **
如果“外群”间的距离太远,则它们可能不可靠,因为它们可能难以可靠比对或已被替换饱和。
2.Midpoint rooting
此方法要求您假设所有序列都以相同的速率进化。
应该谨慎行事,因为这种假设不适用于许多生物学数据集。
**在这种情况下,根位于两个最长分支之间的中点。 **
如果您有taxa在同一时间点未采样到,则需要对该方法进行一些修改,以考虑采样之间的时间间隔。
1.6.5. 置信度
- 困境:没有真实的树关系
推断系统发育是一个固有的不确定性过程,因为我们通常没有比序列更多的信息来指导我们。
有些序列比另一些序列提供更多信息,因此为我们提供了更好的谱系关系估计。
估计我们树的置信度本身就是一个难题。
在生物信息学的其他领域,我们可以检查诸如敏感性和特异性之类的指标,这些指标根据经验已知的真实阳性和阴性实例评估方法的估计或推断结果。
但是,这在系统发育的情况下是不可能的,因为我们没有“已知”祖先序列和系统发育的例子。
- 推断置信度的方法
有几种方法可以用来估计我们对推断树拓扑的置信度:
bootstraps, likelihood and Bayesian approaches
我们以bootstrap为例:
这是通过获取数据集的随机子集来完成的,从每个子集中构建树,并计算每个随机子集中树的各个部分重新生成的频率。 如果group X在每个子集树中都存在,则其confidence为100%;如果仅在三分之二的子集树中找到它,则其confidence为67% .
见下图所示:
通过对序列位点进行有放回的抽样。
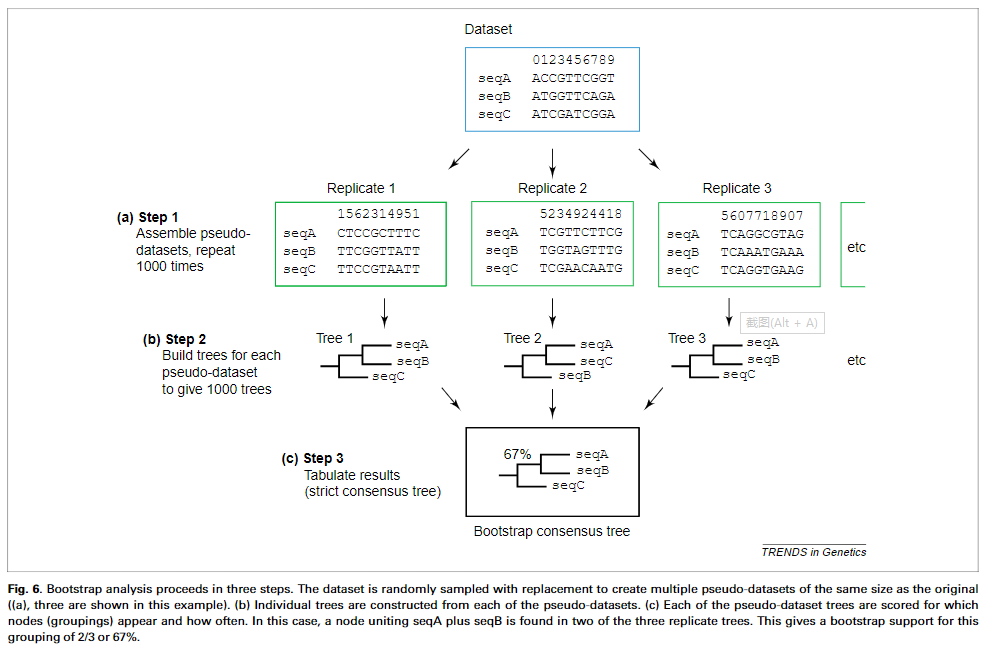
- 如何解读置信度
对树的置信度估计是指它们所显示的内部分支
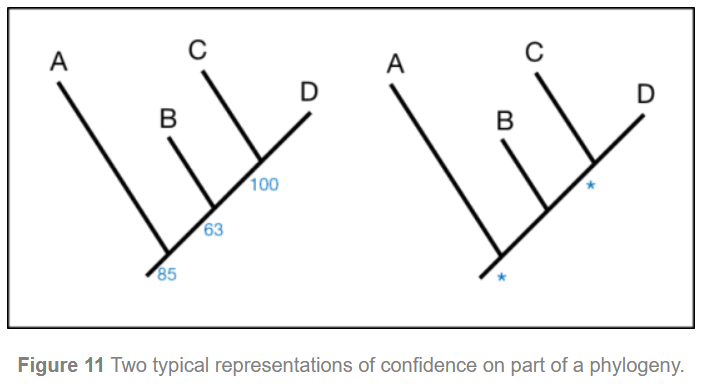
通常,您会在系统发育树上看到bootstrap(或其他置信度估计)值。
例如,在上图中(左),从100次重复中显示了bootstrap值。 有时使用星号(*)来表示引导支持超过80%(或90%)的分支(例如,图11;右)。
在此示例中,引导值100中的63(63%)没有用星号表示,因为它的支持率小于80%。
解释系统发育上的置信度值的确切含义仍是一个争论的话题,但是专家普遍同意,我们可以接受> 80%bootstrap(或90%取决于您的要求!)的分支。只要用合适的进化模型, 被用来估计系统发育。
上图的解读:
(1)“存在一致的(100%bootstrap)支持,即分类单元C和D相互之间的关联比与B之间的关联更为紧密”;
(2)“根据这些数据,不清楚B,C和D是彼此的近亲”。
(1) “there is consistent (100% bootstrap) support that taxa C and D are more closely related to each other than they are to B”;
(2) “from these data it is unclear that B, C and D are each other’s closest relatives”.
1.6.6. Relating distance, rate and time(含对树的理解)
- 进化的速度和时间是混淆的
已经知道,分支的长度会反应遗传改变。
遗传变化是通过替换率和消逝的时间的组合来计算的 :
Genetic change = Evolutionary rate x Divergence time
(substitutions/site) (substitutions/site/year) (years)
每年突变的速率*时间=总突变
这意味着,在没有关于树的任何其他先验信息的情况下,我们不知道下面显示的分支D(图12)是否代表了很久以前采样的不存在或冻结的世系; 进化速度相对较慢的血统; 或两者的某种组合。
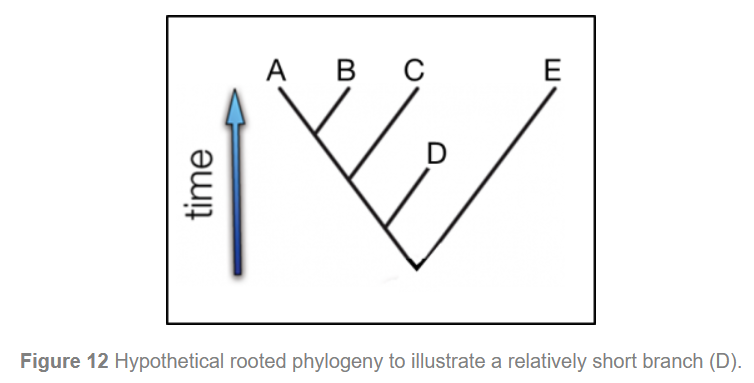
对上图的理解:
一棵树能够推断出真实存在的或者假定的序列的进化关系;
根节点反映了这些序列的公共祖先,这个祖先经过不断发生遗传变化,最终得到了A/B/C/D/E 5个taxa;
而Genetic change = Evolutionary rate x Divergence time
显然,如果符合一个进化模型的解释,遗传变化越大,就代表着经过的时间越多。
因此,本质上突变速率可以看作“分子钟”。
所以,对于上图来说,如果分支越长,代表积累的遗传变化就越多。在相同的进化率下,这意味着经过的时间越长。
节点左边越往上,代表了这个序列对于root节点来讲,经过的突变时间越长。
而中间的一些节点(内节点),通常是一些假定的序列,这些假定的序列可能是两个叶子节点的最近公共祖先;
节点越靠下,越能反映出该序列的突变可能发生在很多年前;
一个特殊的点D:它相对于root节点的分歧并不大,所以,理论上说,有可能是一个已经灭绝的物种或者被冰冻住的物种(这样,遗传改变就在若干年前就停止了); 当然,也可能是进化速度相对缓慢的谱系
这使得你在解释树的时候考虑生物的信息和环境变得非常重要。
如果你足够幸运,知道你的样本被分离的日期,那么有一些方法可以帮助你解析进化的过程。
1.6.7. 一个病毒进化的例子
https://artic.network/how-to-read-a-tree.html
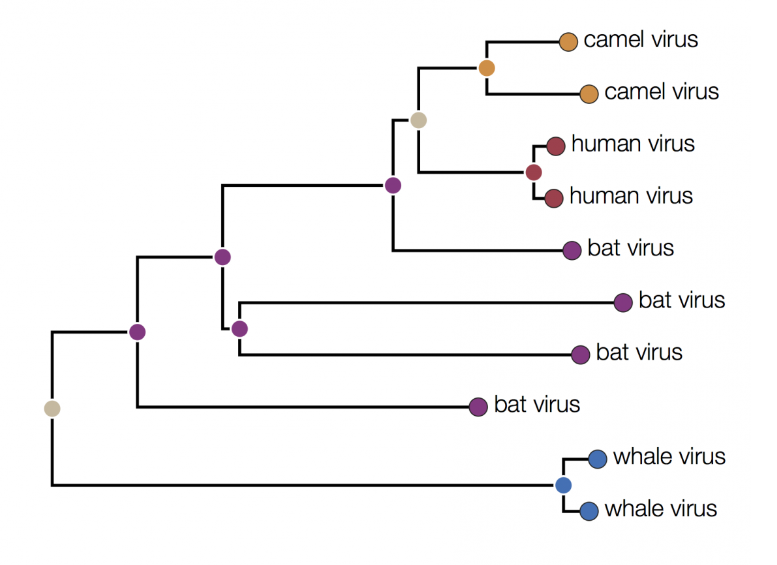
从这幅图中,我们可以推断出蝙蝠病毒是人类以及骆驼病毒的最终来源。
因为最近公共祖先显示, 人类和骆驼病毒的共同祖先存在于所有蝙蝠病毒的多样性中。
1.6.8. 单系群、并系群、多系群
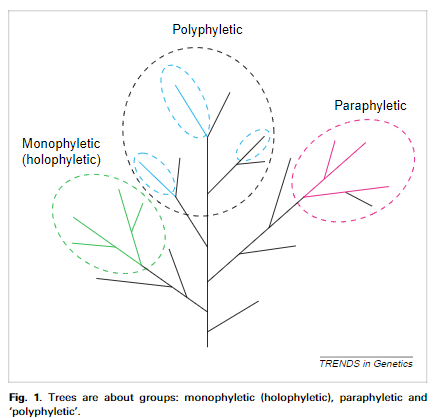
1.单系群 monophyletic
一个节点和由此产生的一切称为一个进化枝或单系群。单系群是一个自然类群。
所有的成员都来自一个独特的共同祖先(相对于树的其余部分),并继承了该祖先的共同特征。
2.并系群 paraphyletic
一个排除其部分后代的群体是一个并并系群。(例如,动物排除人类)。
3.多系群
遥远又相关的大杂烩,也许表面上彼此相似,又或者保留着相似的原始特征,是多系群;
也就是说,根本不是一个group。
1.6.9. 同源性:直系/旁系
Homologues 分为:
直系:orthologues
直系同源的序列因物种形成(speciation)而被区分开(separated):若一个基因原先存在于某个物种,而该物种分化为了两个物种,两个物种中的相同的基因功能未变化,那么新物种中的基因是直系同源的;
直系同源是严格垂直传播的(亲子后代),所以他们的系统发育可以追溯到他们的主谱系 (图B)
旁系:paralogues
旁系同源的序列因基因复制(gene duplication)而被区分开(separated):若生物体中的某个基因被复制了,功能改变了,那么两个副本序列就是旁系同源的。因此,旁系同源基因存在于同一个物种。直系同源的一对序列称为直系同源体(orthologs),旁系同源的一对序列称为旁系同源体(paralogs)。
旁系同源是指多基因族中的成员,如果使用旁系同源基因构建发生树,可能会有点麻烦(如果某个物种丢失了其中一个拷贝,会产生误解,如图C)。
但是,如果您的树中有paralogues的所有拷贝,那么就没问题了。 (图B)
例子:
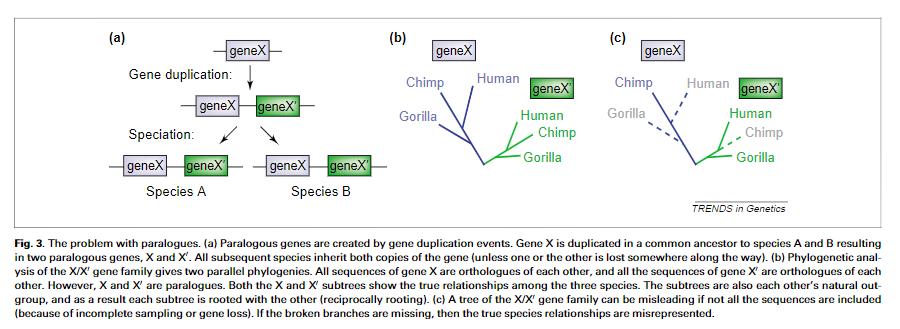
旁系进化树问题。
(a)同源基因是由基因复制事件产生的。 基因X在共同祖先中复制到物种A和B,产生两个旁系基因X和X0。 随后的所有物种都继承了该基因的两个拷贝(除非一个或另一个在此过程中某个地方丢失)。
(b)X / X0基因家族的系统发育分析提供了两个平行的系统发育史。 X基因的所有序列都是彼此的直向同源物,X0基因的所有序列都是彼此的直系同源物。 但是,X和X0是旁系同源。 X和X0子树都显示了这三个物种之间的真实关系。 子树也是彼此的自然组合,因此,每个子树都以彼此为根(相互生根)。
(c)如果未包含所有序列,则X / X0gene家族的树可能会引起误解(由于采样不完整或基因丢失)。 如果缺少折断的分支,则真实的物种关系会被误解。
1.7. 树的其他展示方式
相同的拓扑可以用许多不同的方法绘制。下面的图显示了一些最常见的格式。
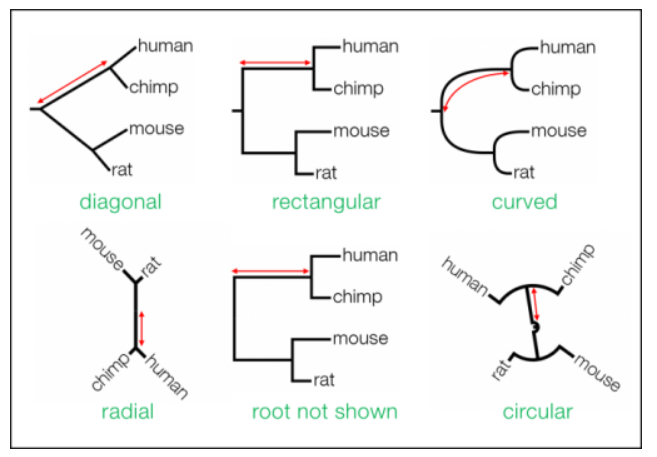
Figure 13 Alternative representations of the same topology.
Red lines indicate the same branch in each representation.
Trees can be rotated on the page and still depict the same tree.
NB: The trees are not drawn to the same scale.
diagonal和 rectangular 格式最常用于发布。
矩形格式的优点是人眼易于快速评估相对分支长度。 但是,您应注意,图中的水平线对应了分支长度,垂直的线的长度没有意义。
curved格式经常用在review论文中来表示系统发育的摘要,因为这类论文对精确了解分支长度可能不太重要。
Radial 格式通常用于显示 unrooted的树,而 circular 格式通常用于表示具有许多taxa的大型系统发育(简单原因是它很容易适合打印页面)。
1.8. 解释亲缘模式
https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/what-phylogeny/interpreting-patterns-relatedness
进阶(一些常见的错误解读): https://evolution.berkeley.edu/evolibrary/misconceptions_faq.php#c1
解释进化内容中的亲缘模式的关键是:
追溯到树中的taxa所共享的LCA。 (看下面第三点解释可以理解这句话)
下面我们将探索脊椎动物系统发育的例子,看看我们如何描述进化关系:
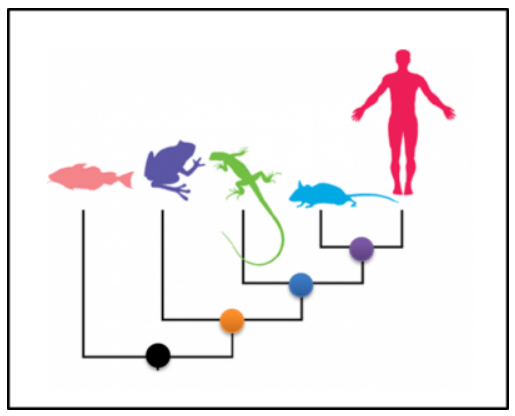
脊椎动物的系统发育,祖先的节用彩色的点突出
1.“人类(红色)与老鼠(蓝色)的亲缘关系比蜥蜴(绿色)的亲缘关系更近。”这是因为人类与老鼠(祖先=紫色斑点)的共同祖先比与蜥蜴(祖先=深蓝色斑点)的共同祖先更近。
2.“青蛙(紫色)与蜥蜴(绿色)的联系比它们与鱼类(粉色)的联系更紧密。”
这是因为青蛙与蜥蜴(鱼=黑斑)的最近公共祖先比,与与鱼的公共祖先最近(祖先=橙色斑点)。”
3.鱼(粉红色)与小鼠(亮蓝色)的关系同青蛙(紫色)的关系相同。” 这不太直观,但是如果您追溯到LCA,您会明白为什么:老鼠和青蛙与鱼类分别具有相同的共同祖先(黑点)。
因此,这两种物种与鱼类的关系都同样紧密。
4.以上3点解释都依赖于根的位置
这些解释依赖于我们的树的根,因为根需要定义进化的方向,因此在该方向上,时间中哪些节点是“最近的”。
These interpretations rely on our tree being rooted because the root is needed to define the direction of evolution and therefore what is ‘more recent’ in evolutionary time.
如果我们旋转树枝来改变树的拓扑结构,那么树仍然具有相同的生物学意义和进化关系。
1.9. 构建进化树的流程
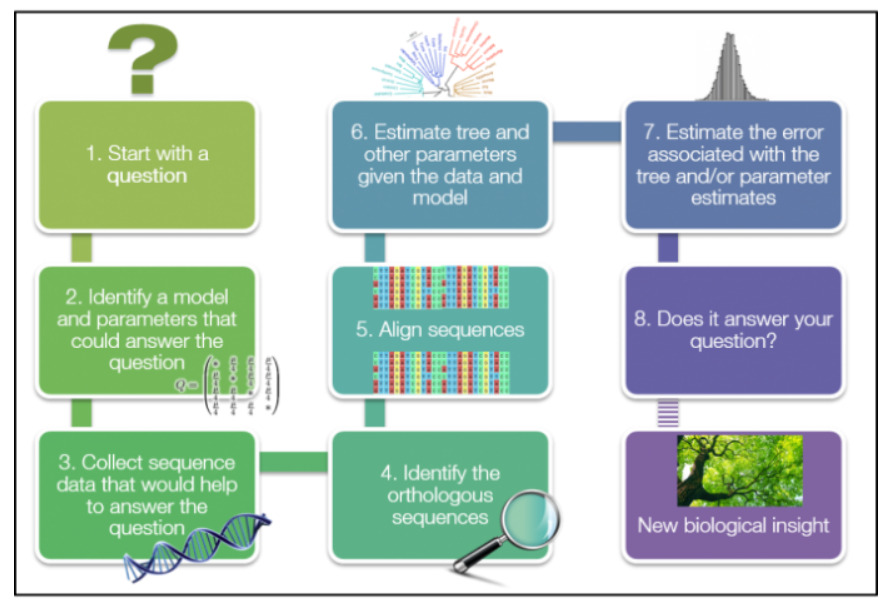
1.审视生物学问题
2.选择模型以及参数
3.收集序列
4.发现直系同源序列
5.比对
6.推断树结构。
7.估计树的错误
8.回顾是否解决了生物学问题
9.发现新insight
1.9.1. 收集序列
1.9.2. 多序列比对
‘progressive sequence alignment’
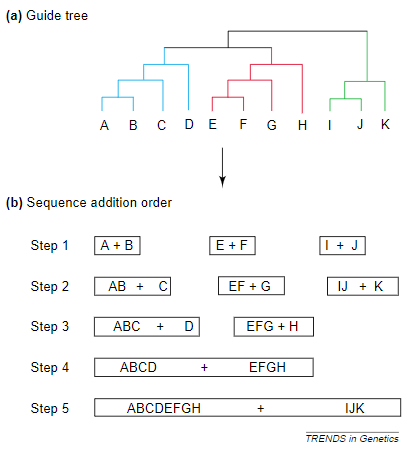
现在,大多数多序列比对都是通过一种称为渐进序列比对的方法来构建的。
这种方法逐步建立比对,从最相似的序列开始,逐步增加更不相似的序列。
具体:
1.首先构建一颗粗糙的guide tree
2.guide tree接下来决定了逐步增加的序列顺序。
渐进序列比对的基本原则是“一旦有gap,总有gap”。 gap只能添加或放大,不能移动或删除。
1.10. EMBL-EMI的系统发生学资源
Ensembl:
是脊椎动物基因组数据的巨大资源,您可以(其中包括)下载核苷酸序列数据并运行BLAST搜索以告知鉴定同源序列。
对于许多基因,您还可以下载我们已经确定的直系同源序列,以及我们已经预先计算的种间比对。
为此,只需搜索您喜欢的基因(例如ADH1B),然后在比较基因组菜单中浏览“基因组比对”选项。
Ensembl基因组:
将Ensembl扩展到生命之树上,使细菌,植物,真菌,原生生物和后生动物的基因组数据公开可用。
这包括预先计算的比对和直向同源物。
Ensembl compara:
为预先计算的系统进化提供可视化和下载。有关更多信息,请参见文档页面。
ClustalW2系统发育:
是一个基本的工具,估计进化树从多个序列比对。它使用了邻接方法和一个非常简单的序列进化模型的选项(Jukes和Cantor, 1969)。
1.11. 资源推荐
https://artic.network/how-to-read-a-tree.html
https://www.ebi.ac.uk/training/online/course/introduction-phylogenetics/
Phylogeny for the faint of heart:a tutorial
Molecular phylogenetics: principles and practice
上一篇: 扩增子序列变异为何取代OTU
下一篇: 多元高斯分布:概念、参数估计与生信中的应用