扩增子序列变异为何取代OTU
本文翻译自
题目:Exact sequence variants should replace operational taxonomic units in marker-gene data analysis
杂志:The ISME Journal(2017) 11, 2639–2643
作者:Benjamin J Callahan, Paul J McMurdie and Susan P Holmes
介绍
扩增子序列变异(ASV),可以准确解析单核苷酸水平的差异。
ASV的概况:the sequence is the label itself.
reproducible,reusable,comprehensive,better resolution and accuracy
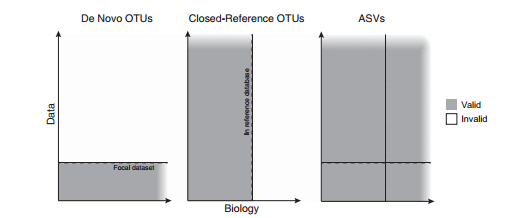
x轴代表存在于测序基因座的生物学变异。
y轴代表从该基因座产生的所有扩增子数据以及所有将来可能产生的数据。
阴影代表有效性。
该图充分反映了各种方法的优缺点。
不同方法的优缺点
De novo OTU方法:
1.在两个不同的数据集中定义的OTU不能比较。
2.在进行de nove方法的时候,由于不同的数据不具备可比性,因此即使在同一项研究下,也必须把所有样本pool在一起,潜在的比较次数与总测序呈二次方关系,耗时最长。
3.不利于再现研究中的结论。
Closed-reference OTUs:
1.当两个数据集使用的是同一个参考库的时候,该两个数据集是可比较的。
2.容易丢失那些参考数据库中不存在的变异。
3.diversity分析存在bias:对于那些比对不上参考序列的序列,如果直接扔掉,会影响diversity的估计。(这与样本来源有关,如果样本来自非常规渠道,很有可能丢失大量的序列)
4.无法保证注释到的OTU一定是样本中存在的OTU:注视到OTU的条件是至少存在一个测序reads与参考序列足够相似。然而,参考序列是数据库中选取的代表序列,并不一定会在自己的样本中观测到。如果使用了代表序列进行分析,那么在下游时可能会产生误导性的结果。
5.OTU的注释严重依赖于参考数据库,而与样本本身序列无关。因此,不同数据库注释出来的数据不具备可比性。如果想要在不同时间节点注释的结果具备可比性,当且仅当满足下面条件中的一条:1.数据库是静态的;2.每当数据库升级后,重新生成OTU表。(现实是很难满足,这意味着:在2021年的结果可能与现在得到的结果不一致)
ASV
ASV通过从头开始的过程进行推断,其中生物学序列与错误区分开,原理在于相比于测序错误碱基,生物学序列变异的期望更有可能被重复观测到(思考:DADA2 partition算法的原理)
1.因此,ASV最少需要以一个样本为单位进行推断。因此,可以以样本为单位,进行并行化的样本组成推断。这就使得计算时间得以扩展线性变化,内存需求保持平稳,随着样本数量的增加,ASV可以从任意大数据集中进行推断。
2.从不同的研究中独立推断的ASV,是具备可比较性的,因为ASV本身就代表被测生物的DNA序列。
3.方便进行荟萃分析。
局限:
ASV并不能解决所有问题:
1.扩增的基因座在一个基因组上存在多拷贝的情况,从而存在多个ASV。
2.即便核糖体序列变异完全相同,仍旧无法保证生态学上的连贯性、甚至单系群。( no guarantee of ecological coherence or even monophyly among genomes )
monophyly:单系群
In cladistics, a monophyletic group, or clade, is a group of organisms that consists of all the descendants of a common ancestor (or more precisely ancestral population). Monophyletic groups are typically characterised by shared derived characteristics (synapomorphies), which distinguish organisms in the clade from other organisms. The arrangement of the members of a monophyletic group is called a monophyly.
3.实践中,合并不同研究的ASV表格是有局限的。 来自相同的基因座的ASV表格可以直接合并(这意味着,研究之间必须采用相同的引物)。否则,需要人为取overlapping的基因区域进行ASV表格合并。
4.不同的ASV算法可能存在批次效应。
上一篇: 详解DADA2算法原理
下一篇: Introduction to Phylogenetic Tree